医薬品情報管理学[3]
医薬品情報21
古泉秀夫
| 情報管理の基本 |
1] 守備範囲の決定
『情報は限りなく空間を占有する』。
情報管理業務を行う上で、最も重要な課題の一つは、蒐集した情報の保管をどうするかということである。病院における医薬品情報管理室を考えた場合、病院より大きな薬局は存在し得ないのと同様、薬局より大きな医薬品情報管理室は存在しない。薬局は病院にとって一つの機関であり、医薬品情報管理室は、1 機関における一つの機能でしかない。従って、膨大な情報量を蒐集したとしても、保管する場所がなければ、それは単なる紙屑を蒐集したに過ぎないということである。
例えば5種類の月刊雑誌を購入しているとして、12冊×5=60冊の雑誌が年間配本されることになる。これだけの雑誌の数を数年にわたって保存するだけで、広大な空間を占有されることになるのである。更に5種類の雑誌で全ての情報を網羅することは困難である。いずれにしろ医療・医薬品に関する情報は際限なく作成される。その全ての情報を1情報管理室で蒐集・管理することは不可能ということである。
情報の奔流の中に三連の水車を設置し、情報を汲み上げたところで、全ての情報を汲み上げることは出来ない。まして情報の奔流をせき止めたとすれば、大洪水を起こし、情報の管理は不可能になる。
従って、1医療機関 の医薬品情報管理室で実行できる情報管理の守備範囲には、自ずから限界があるということである。また、そのことを前提に、業務の推進を図らなければ、情報管理室そのものの機能を停止させかねない。更に直接的な診療報酬の対象とならない情報管理室に、多くの薬剤師を配置するということは、資本主義経済の原則である投下資本に見合う収益性を考えた場合、不可能である。医薬品情報管理室が機能し、新薬採用に伴う致命的な相互作用を阻止したとしても、あるいは医薬品の誤用による事故の際に、情報の提供により致死的状況を防いだとしても、収益性が無いという大前提の下では、人員配置要求も説得力がないということである。

図5.情報蒐集モデル例-朝倉三連水車(写真提供:倉田稔氏)
2]添付文書
医薬品の場合、各個別の商品に、その薬に関する情報を添付することが、法的に定められている。従前、『能書(効能書)』、『説明書』、『巻紙』等といわれていたが、法的には『添付文書』である。
添付文書等の記載事項については、薬事法第52条に『医薬品は、これに添付する文書又はその容器若しくは被包に、次の各号に掲げる事項が記載されていなければならない。ただし、厚生省令で別段の定めをしたときは、この限りではない。』と定められている。
- 用法、用量その他使用及び取扱い上の注意
- 日本薬局方に収められている医薬品にあっては、日本薬局方においてこれに添付する文書又はその容器若しくは被包に記載するよう定められている事項
- 第42条第1項の規定によりその基準が定められた医薬品にあっては、その基準においてこれに添付する文書又はその容器若しくは被包に記載するように定められた事項
- 前各号に掲げるもののほか、厚生省令で定める事項
また、添付文書等への記載禁止事項については、第54条『医薬品は、これに添付する文書、その医薬品又はその容器若しくは被包(内袋を含む。)に、次の各号に掲げる事項が記載されていてはならない。』と定められている。
- 当該医薬品に関して虚偽又は誤解を招くおそれのある事項
- 第14条(第23条)において準用する場合を含む。以下同じ。)又は第19条の2の規定による承認を受けていない効能又は効果(第14条第1項の規定により厚生大臣がその基準を定めて指定した医薬品にあってはその基準において定められた効能又は効果を除く。)
- 保健衛生上危険がある用法、用量又は使用期限
*第14条:医薬品等の製造の承認(14-2:前項の承認は、申請に係わる医薬品、医薬部外品等の名称、成分、分量、構造、用法、用量、使用方法、効能、効果、性能、副作用等を審査して行うものとし………)。
*第19条の2:外国製造医薬品等の製造の承認
以上に見るまでもなく、医薬品そのものは厚生大臣の許可なしには製造できないものであり、医薬品の使用は、製品に添付されている添付文書によることが当然ということである。つまり医薬品に関する情報の基本は、法律で定められた『添付文書』によるということである。
添付文書は、製薬企業が医薬品として開発すべき『物』を選択して基礎実験を開始し、各段階の臨床試験を実施する過程で蒐集した多くの情報を圧縮して収載したものである。ただし、臨床治験段階の医薬品は、ある意味で“選別した患者”を対象としたものであり、その薬剤の本質的な部分で、一部不明のまま市場に出されるという問題点を抱えている。例えば、
- 『腎障害のある患者への投与』
- 『肝障害のある患者への投与』
- 『高齢者・幼児・小児への投与量』
- 『食物・嗜好品と薬の相互作用』
- 『母乳中移行』
- 『配合変化』
等の情報は、臨床治験段階では、種々の困難があって、簡単には入手出来ない情報だといえる。
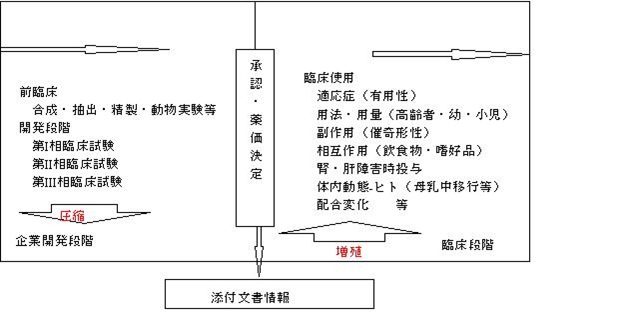
図6.添付文書の情報内容
これらの情報の多くは、臨床現場で医薬品として使用されて初めて確認できる情報であり、時には臨床治験段階では予測されなかった使用によって、初めて入手される情報なのである。
3]添付文書情報の重み
添付文書情報の重要性は、単に薬事法上に定められた文書というだけではなく、その薬を使用する上で、患者の安全性を確保するための最低限の情報を提供するものであるということである。
『医薬品の添付文書の記載事項は、当該医薬品の危険性(副作用等)につき最も高度な情報を有している製造業者又は輸入販売業者が投与を受ける患者の安全を確保するために、これを使用する医師等に対して必要な情報を提供する目的で記載するものであるから、医師が医薬品を使用するにあたって添付文書に記載された使用上の注意義務に従わず、それによって医療事故が発生した場合には、これに従わなかったことにつき特段の合理的理由がない限り、当該医師の過失が推定されるものというべきである』
1996年(平成8年)1月23日、ペルカミンSによる腰椎麻酔に関連した医療事故に対する最高裁判決中に見られる判断である。
1972年(昭和47年)に0.3%-ペルカミンSの添付文書中に、血圧対策として『麻酔剤注入前に1回、注入後は10-15分まで2分間隔で血圧を測定する』の記載が追記された。しかし、この当時の医療上の慣行では『5分間隔で血圧を測定する』というのが一般開業医の常識とされており、1審及び2 審では、これを『当時の医療水準を基準とする限り、過失とはいえない』として患者側が敗訴していた。
これに対し最高裁は『医師が医薬品を使用するにあたって、医薬品の添付文書に記載された使用上の注意事項に従わなかったことについて、特段の合理的理由がない限り、医師の過失が推定される』として、担当医師には『2分ごとに血圧測定を行わなかった過失があり、この過失と患者の脳機能低下症発症との間には因果関係がある』として、原判決を一部破棄し、破棄部分を原審に差し戻しした(その後、高裁で和解)。
[註]
医療水準;一般的には『診療当時の臨床医学の実践における医療水準は、全国一律に絶対的な基準として考えるものではなく、診療にあたる当該医師の専門分野、所属する診療機関の性格、その所在する地域の医療環境の特性等の諸般の事情を考慮して決せられるべきものであるが、医療水準は、医師の注意義務の基準(規範)となるものであるから、平均的医師が現に行っている医療慣行とは必ずしも一致するものではなく、医師が医療慣行に従った医療行為を行ったからといって、医療水準に従って注意義務を尽くしたと、直ちにいうことは出来ない』
添付文書は、患者の安全性を確保するため、その医薬品の効能、危険性等について最も情報を把握している製造業者等にこれを使用する医師等に対し、必要な情報を提供すべく薬事法第52条により義務付けた規定である。
従って、医師が医薬品を使用するに際には、特段の合理的理由がない限り、原則として添付文書の記載事項に従う義務があるとするのは自明のことであり、添付文書の注意事項に高度の規範性を認めたこの最高裁判決は、画期的な判断を示したということが出来る。更に1985年(昭和60年)4月9日の『チトクロームC』の使用方法に関する最高裁判決は、『添付文書に十分な記載がされていなくとも、それに付加し、適切な問診が不可欠であることが臨床医の間で一般的に認められている場合、これに従うべきである』としている。
添付文書に関するこの判決は、最高裁の判決であり、今後の医療訴訟の際の判断基準とされるものであり、医薬品を使用する際、『添付文書は最低限守るべき規範』であるとすることが出きる。
1)増田聖子:医薬品の添付文書と医師の注意義務;臨床医,27(4):540-543(2001)